I. Introduction
This guidance is intended to help pharmaceutical manufacturers meet the requirements in the US FDA’S current good manufacturing practice (CGMP) regulations (2l CFR parts 210 and 211) when manufacturing sterile drug and biological products using aseptic processing. This guidance replaces the 1987 Industry Guideline on Sterile Drug Products Produced by Aseptic Processing (Aseptic Processing Guideline). This revision updates and clarifies the 1987 guidance.
For sterile drug products subject to a new or abbreviated drug application (NDA or ANDA) or a biologic license application (BLA), this guidance document should be read in conjunction with the guidance on the content of sterile drug applications entitled Guideline for the Submission of Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products (Submission Guidance). The Submission Guidance describes the types of information and data that should be included in drug applications to demonstrate the efficacy of a manufacturer’s sterilization process. This guidance compliments the Submission Guidance by describing procedures and practices that will help enable a sterile drug manufacturing facility to meet CGMP requirements relating, for example, to facility design, equipment suitability, process validation, and quality control.
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the US FDA’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in fda’s guidances means that something is suggested or recommended, but not required.
The text boxes included in this guidance include specific sections of parts 210 and 211 of the Code of Federal Regulations (CFR), which address current good manufacturing practice for drugs pharmaceuticals . The intent of including these quotes in the text boxes is to aid the reader by providing a portion of an applicable regulation being addressed in the guidance. The quotes included in the text boxes are not intended to be exhaustive. Readers of this document should reference the complete CFR to ensure that they have complied, in full, with all relevant sections of the regulations.
II. Background
This section describes briefly both the pharmaceutical regulatory and technical reasons why the Agency is developing this guidance document.
A. Regulatory Framework
This guidance pertains to current good manufacturing practice (CGMP) regulations (21 CFR parts 210 and 211) when manufacturing sterile drug and biological products using aseptic processing. Although the focus of this guidance is on CGMPs in 21 CFR 210 and 211, supplementary requirements for biological products are in 21 CFR 600-680. For biological products regulated under 21 CFR parts 600 through 680, §§ 210.2(a) and 211.1(b) provide that where it is impossible to comply with the applicable regulations in both parts 600 through 680 and parts 210 and 211, the regulation specifically applicable to the drug product in question shall supercede the more general regulations.
B. Technical Framework
There are basic differences between the production of sterile drug pharmaceutical products using aseptic processing and production using terminal sterilization.
Terminal sterilization usually involves filling and sealing product containers under high-quality environmental conditions. Products are filled and sealed in this type of environment to minimize the microbial and particulate content of the in-process product and to help ensure that the subsequent sterilization process is successful. In most cases, the product, container, and closure have low bioburden, but they are not sterile. The product in its final container is then subjected to a sterilization process such as heat or irradiation.
In an aseptic process, the drug product, container, and closure are first subjected to sterilization methods separately, as appropriate, and then brought together. Because there is no process to sterilize the product in its final container, it is critical that containers be filled and sealed in an extremely high-quality environment. Aseptic processing involves more variables than terminal sterilization. Before aseptic assembly into a final product, the individual parts of the final product are generally subjected to various sterilization processes. For example, glass containers are subjected to dry heat; rubber closures are subjected to moist heat; and liquid dosage forms are subjected to filtration. Each of these manufacturing processes requires validation and control. Each process could introduce an error that ultimately could lead to the distribution of a contaminated product. Any manual or mechanical manipulation of the sterilized drug, components, containers, or closures prior to or during aseptic assembly poses the risk of contamination and thus necessitates careful control. A terminally sterilized drug product, on the other hand, undergoes final sterilization in a sealed container, thus limiting the possibility of error.[3]
Sterile drug manufacturers should have a keen awareness of the public health implications of distributing a nonsterile product. Poor CGMP conditions at a manufacturing facility can ultimately pose a life-threatening health risk to a patient.
III. Scope
This guidance document discusses selected issues and does not address all aspects of aseptic processing. For example, the guidance addresses primarily finished drug product CGMP issues while only limited information is provided regarding upstream bulk processing steps. This guidance updates the 1987 Aseptic Processing Guideline primarily with respect to personnel qualification, cleanroom design, process design, quality control, environmental monitoring, and review of production records. The use of isolators for aseptic processing is also discussed.
Although this guidance document discusses CGMP issues relating to the sterilization of components, containers, and closures, terminal sterilization of drug products is not addressed. It is a well-accepted principle that sterile drugs should be manufactured using aseptic processing only when terminal sterilization is not feasible. However, some final packaging may afford some unique and substantial advantage (e.g., some dual-chamber syringes) that would not be possible if terminal sterilization were employed. In such cases, a manufacturer can explore the option of adding adjunct processing steps to increase the level of sterility assurance.
A list of references that may be of value to the reader is included at the conclusion of this document.
IV. BUILDINGS AND FACILITIES
21 CFR 211.42(b) states, in part, that “The flow of components, drug product containers, closures, labeling, in-process materials, and drug products through the building or buildings shall be designed to prevent contamination.”
21 CFR 211.42(c) states, in part, that “Operations shall be performed within specifically defined areas of adequate size. There shall be separate or defined areas or such other control systems for the firm’s operations as are necessary to prevent contamination or mixups during the course of the following procedures (10) Aseptic processing, which includes as appropriate: (i) Floors, walls, and ceilings of smooth, hard surfaces that are easily cleanable; (ii) Temperature and humidity controls; (iii) An air supply filtered through high-efficiency particulate air filters under positive pressure, regardless of whether flow is laminar or nonlaminar; (iv) A system for monitoring environmental conditions; (v) A system for cleaning and disinfecting the room and equipment to produce aseptic conditions; (vi) A system for maintaining any equipment used to control the aseptic conditions.”
21 CFR 211.46(b) states that “Equipment for adequate control over air pressure, micro-organisms, dust, humidity, and temperature shall be provided when appropriate for the manufacture, processing, packing, or holding of a drug product.”
21 CFR 211.46(c) states, in part, that “Air filtration systems, including prefilters and particulate matter air filters, shall be used when appropriate on air supplies to production areas
21 CFR 211.63 states that “Equipment used in the manufacture, processing, packing, or holding of a drug product shall be of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance.”
21 CFR 211.65(a) states that “Equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.”
21 CFR 211.67(a) states that “Equipment and utensils shall be cleaned, maintained, and sanitized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.”
21 CFR 211.113(b) states that “Appropriate written procedures, designed to prevent microbiological contamination of drug products purporting to be sterile, shall be established and followed. Such procedures shall include validation of any sterilization process.”
As provided for in the regulations, separate or defined areas of operation in an aseptic processing facility should be appropriately controlled to attain different degrees of air quality depending on the nature of the operation. Design of a given area involves satisfying microbiological and particle criteria as defined by the equipment, components, and products exposed, as well as the operational activities conducted in the area.
Clean area control parameters should be supported by microbiological and particle data obtained during qualification studies. Initial cleanroom qualification includes, in part, an assessment of air quality under as-built, static conditions. It is important for area qualification and classification to place most emphasis on data generated under dynamic conditions (i.e., with personnel present, equipment in place, and operations ongoing). An adequate aseptic processing facility monitoring program also will assess conformance with specified clean area classifications under dynamic conditions on a routine basis.
The following table summarizes clean area air classifications and recommended action levels of microbiological quality (Ref. 1).
TABLE 1- Air Classificationsa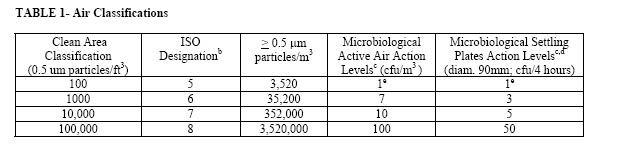
Action Lvel CFU /4 HRS*
Clean Area Classification
(0.5 um particles/ft3) ISO
Designationb > 0.5 m particles/m3 Microbiological Active Air Action Levelsc (cfu/m3 ) Microbiological Settling Plates Action Levelsc,d (diam. 90mm; cfu/4 hours)
100 5 3,520 1e 1e
1000 6 35,200 7 3
10,000 7 352,000 10 5
100,000 8 3,520,000 100 50
a- All classifications based on data measured in the vicinity of exposed materials/articles during periods of activity.
b- ISO 14644-1 designations provide uniform particle concentration values for cleanrooms in multiple industries. An ISO 5 particle concentration is equal to Class 100 and approximately equals EU Grade A.
c- Values represent recommended levels of environmental quality. You may find it appropriate to establish alternate microbiological action levels due to the nature of the operation or method of analysis.
d- The additional use of settling plates is optional.
e- Samples from Class 100 (ISO 5) environments should normally yield no microbiological contaminants.
Two clean areas are of particular importance to sterile pharmaceutical drug product quality: the critical area and the supporting clean areas associated with it.
A. Critical Area – Class 100 (ISO 5)
A critical area is one in which the sterilized drug product, containers, and closures are exposed to environmental conditions that must be designed to maintain pharmaceutical product sterility (§ 211.42(c)(10)). Activities conducted in such areas include manipulations (e.g., aseptic connections, sterile ingredient additions) of sterile materials prior to and during filling and closing operations.
This area is critical because an exposed product is vulnerable to contamination and will not be subsequently sterilized in its immediate container. To maintain product sterility, it is essential that the environment in which aseptic operations (e.g., equipment setup, filling) are conducted be controlled and maintained at an appropriate quality. One aspect of environmental quality is the particle content of the air. Particles are significant because they can enter a product as an extraneous contaminant, and can also contaminate it biologically by acting as a vehicle for microorganisms (Ref. 2). Appropriately designed air handling systems minimize particle content of a critical area.
Air in the immediate proximity of exposed sterilized containers/closures and filling/closing operations would be of appropriate particle quality when it has a per-cubic-meter particle count of no more than 3520 in a size range of 0.5 m and larger when counted at representative locations normally not more than 1 foot away from the work site, within the airflow, and during filling/closing operations. This level of air cleanliness is also known as Class 100 (ISO 5).
US FDA recommend that measurements to confirm air cleanliness in critical areas be taken at sites where there is most potential risk to the exposed sterilized product, containers, and closures. The particle counting probe should be placed in an orientation demonstrated to obtain a meaningful sample. Regular monitoring should be performed during each production shift. US FDA recommend conducting nonviable particle monitoring with a remote counting system. These systems are capable of collecting more comprehensive data and are generally less invasive than portable particle counters. See Section X.E. for additional guidance on particle monitoring.
Some operations can generate high levels of product (e.g., powder) particles that, by their nature, do not pose a risk of product contamination. It may not, in these cases, be feasible to measure air quality within the one-foot distance and still differentiate background levels of particles from air contaminants. In these instances, air can be sampled in a manner that, to the extent possible, characterizes the true level of extrinsic particle contamination to which the product is exposed. Initial qualification of the area under dynamic conditions without the actual filling function provides some baseline information on the non-product particle generation of the operation.
HEPA-filtered air should be supplied in critical areas at a velocity sufficient to sweep particles away from the filling/closing area and maintain unidirectional airflow during operations. The velocity parameters established for each processing line should be justified and appropriate to maintain unidirectional airflow and air quality under dynamic conditions within the critical area (Ref. 3).[5]
Proper design and control prevents turbulence and stagnant air in the critical area. Once relevant parameters are established, it is crucial that airflow patterns be evaluated for turbulence or eddy currents that can act as a channel or reservoir for air contaminants (e.g., from an adjoining lower classified area). In situ air pattern analysis should be conducted at the critical area to demonstrate unidirectional airflow and sweeping action over and away from the product under dynamic conditions. The studies should be well documented with written conclusions, and include evaluation of the impact of aseptic manipulations (e.g., interventions) and equipment design. Videotape or other recording mechanisms have been found to be useful aides in assessing airflow initially as well as facilitating evaluation of subsequent equipment configuration changes. It is important to note that even successfully qualified systems can be compromised by poor operational, maintenance, or personnel practices.
Air monitoring samples of critical areas should normally yield no microbiological contaminants. US FDA recommend affording appropriate investigative attention to contamination occurrences in this environment.
B. Supporting Clean Areas
Supporting clean areas can have various classifications and functions. Many support areas function as zones in which nonsterile components, formulated products, in-process materials, equipment, and container/closures are prepared, held, or transferred. These environments are soundly designed when they minimize the level of particle contaminants in the final product and control the microbiological content (bioburden) of articles and components that are subsequently sterilized.
The nature of the activities conducted in a supporting clean area determines its classification. FDA recommends that the area immediately adjacent to the aseptic processing line meet, at a minimum, Class 10,000 (ISO 7) standards (see Table 1) under dynamic conditions. Manufacturers can also classify this area as Class 1,000 (ISO 6) or maintain the entire aseptic filling room at Class 100 (ISO 5). An area classified at a Class 100,000 (ISO 8) air cleanliness level is appropriate for less critical activities (e.g., equipment cleaning).
C. Clean Area Separation
An essential part of contamination prevention is the adequate separation of areas of operation. To maintain air quality, it is important to achieve a proper airflow from areas of higher cleanliness to adjacent less clean areas. It is vital for rooms of higher air cleanliness to have a substantial positive pressure differential relative to adjacent rooms of lower air cleanliness. For example, a positive pressure differential of at least 10-15 Pascals (Pa)[6] should be maintained between adjacent rooms of differing classification (with doors closed). When doors are open, outward airflow should be sufficient to minimize ingress of contamination, and it is critical that the time a door can remain ajar be strictly controlled (Ref. 4).
In some cases, the aseptic processing room and adjacent cleanrooms have the same classification. Maintaining a pressure differential (with doors closed) between the aseptic processing room and these adjacent rooms can provide beneficial separation. In any facility designed with an unclassified room adjacent to the aseptic processing room, a substantial overpressure (e.g., at least 12.5 Pa) from the aseptic processing room should be maintained at all times to prevent contamination. If this pressure differential drops below the minimum limit, it is important that the environmental quality of the aseptic processing room be restored and confirmed.
The Agency recommends that pressure differentials between cleanrooms be monitored continuously throughout each shift and frequently recorded. All alarms should be documented and deviations from established limits should be investigated.
Air change rate is another important cleanroom design parameter. For Class 100,000 (ISO 8) supporting rooms, airflow sufficient to achieve at least 20 air changes per hour is typically acceptable. Significantly higher air change rates are normally needed for Class 10,000 and Class 100 areas.
A suitable facility monitoring system will rapidly detect atypical changes that can compromise the facility’s environment. An effective system facilitates restoration of operating conditions to established, qualified levels before reaching action levels. For example, pressure differential specifications should enable prompt detection (i.e., alarms) of an emerging low pressure problem to preclude ingress of unclassified air into a classified room.
D. Air Filtration
1. Membrane
A compressed gas should be of appropriate purity (e.g., free from oil) and its microbiological and particle quality after filtration should be equal to or better than that of the air in the environment into which the gas is introduced. Compressed gases such as air, nitrogen, and carbon dioxide are often used in cleanrooms and are frequently employed in purging or overlaying.
Membrane filters can be used to filter a compressed gas to meet an appropriate high-quality standard. These filters are often used to produce a sterile compressed gas to conduct operations involving sterile materials, such as components and equipment. For example, US FDA recommend that sterile membrane filters be used for autoclave air lines, lyophilizer vacuum breaks, and tanks containing sterilized materials. Sterilized holding tanks and any contained liquids should be held under positive pressure or appropriately sealed to prevent microbial contamination. Safeguards should be in place to prevent a pressure change that can result in contamination due to back flow of nonsterile air or liquid.
Gas filters (including vent filters) should be dry. Condensate on a gas filter can cause blockage during use or allow for the growth of microorganisms. Use of hydrophobic filters, as well as application of heat to these filters where appropriate, prevents problematic moisture residues. US FDA recommend that filters that serve as sterile boundaries or supply sterile gases that can affect product be integrity tested upon installation and periodically thereafter (e.g., end of use). Integrity tests are also recommended after activities that may damage the filter. Integrity test failures should be investigated, and filters should be replaced at appropriate, defined intervals.
2. High-Efficiency Particulate Air (HEPA)
HEPA filter integrity should be maintained to ensure aseptic conditions. Leak testing should be performed at installation to detect integrity breaches around the sealing gaskets, through the frames, or through various points on the filter media. Thereafter, leak tests should be performed at suitable time intervals for HEPA filters in the aseptic processing facility. For example, such testing should be performed twice a year for the aseptic processing room. Additional testing may be appropriate when air quality is found to be unacceptable, facility renovations might be the cause of disturbances to ceiling or wall structures, or as part of an investigation into a media fill or drug product sterility failure. Among the filters that should be leak tested are those installed in dry heat depyrogenation tunnels and ovens commonly used to depyrogenate glass vials. Where justified, alternate methods can be used to test HEPA filters in the hot zones of these tunnels and ovens.
Any aerosol used for challenging a HEPA filter should meet specifications for critical physicochemical attributes such as viscosity. Dioctylphthalate (DOP) and poly-alpha-olefin (PAO) are examples of appropriate leak testing aerosols. Some aerosols are problematic because they pose the risk of microbial contamination of the environment being tested. Accordingly, the evaluation of any alternative aerosol involves ensuring it does not promote microbial growth.
There is a major difference between filter leak testing and efficiency testing. An efficiency test is a general test used to determine the rating of the filter. An intact HEPA filter should be capable of retaining at least 99.97 percent of particulates greater than 0.3 nm in diameter.
The purpose of performing regularly scheduled leak tests, on the other hand, is to detect leaks from the filter media, filter frame, or seal. The challenge involves use of a polydispersed aerosol usually composed of particles with a light-scattering mean droplet diameter in the submicron size range, including a sufficient number of particles at approximately 0.3 m. Performing a leak test without introducing a sufficient upstream challenge of particles of known size upstream of the filter is ineffective for detecting leaks. It is important to introduce an aerosol upstream of the filter in a concentration that is appropriate for the accuracy of the aerosol photometer. The leak test should be done in place, and the filter face scanned on the downstream side with an appropriate photometer probe, at a sampling rate of at least one cubic foot per minute. The downstream leakage measured by the probe should then be calculated as a percent of the upstream challenge. An appropriate scan should be conducted on the entire filter face and frame, at a position about one to two inches from the face of the filter. This comprehensive scanning of HEPA filters should be fully documented.
A single probe reading equivalent to 0.01 percent of the upstream challenge would be considered as indicative of a significant leak and calls for replacement of the HEPA filter or, when appropriate, repair in a limited area. A subsequent confirmatory retest should be performed in the area of any repair.
HEPA filter leak testing alone is insufficient to monitor filter performance. It is important to conduct periodic monitoring of filter attributes such as uniformity of velocity across the filter (and relative to adjacent filters). Variations in velocity can cause turbulence that increases the possibility of contamination. Velocities of unidirectional air should be measured 6 inches from the filter face and at a defined distance proximal to the workHEPA filters in the critical area. Velocity monitoring at suitable intervals can provide useful data on the critical area in which aseptic processing is performed. The measurements should correlate to the velocity range established at the time of in situ air pattern analysis studies. HEPA filters should be replaced when nonuniformity of air velocity across an area of the filter is detected or airflow patterns may be adversely affected.
Although contractors often provide these services, drug manufacturers are responsible for ensuring that equipment specifications, test methods, and acceptance criteria are defined, and that these essential certification activities are conducted satisfactorily.
E. Design
Note: The design concepts discussed within this section are not intended to be exhaustive. Other appropriate technologies that achieve increased sterility assurance are also encouraged.
Aseptic processes are designed to minimize exposure of sterile articles to the potential contamination hazards of the manufacturing operation. Limiting the duration of exposure of sterile product elements, providing the highest possible environmental control, optimizing process flow, and designing equipment to prevent entrainment of lower quality air into the Class 100 (ISO 5) clean area are essential to achieving high assurance of sterility .
Both personnel and material flow should be optimized to prevent unnecessary activities that could increase the potential for introducing contaminants to exposed product, container-closures, or the surrounding environment. The layout of equipment should provide for ergonomics that optimize comfort and movement of operators. The number of personnel in an aseptic processing room should be minimized. The flow of personnel should be designed to limit the frequency with which entries and exits are made to and from an aseptic processing room and, most significant, its critical area. Regarding the latter, the number of transfers into the critical area of a traditional cleanroom, or an isolator, should be minimized. To prevent changes in air currents that introduce lower quality air, movement adjacent to the critical area should be appropriately restricted.
Any intervention or stoppage during an aseptic process can increase the risk of contamination. The design of equipment used in aseptic processing should limit the number and complexity of aseptic interventions by personnel. For example, personnel intervention can be reduced by integrating an on-line weight check device, thus eliminating a repeated manual activity within the critical area. Rather than performing an aseptic connection, sterilizing the preassembled connection using sterilize-in-place (SIP) technology also can eliminate a significant aseptic manipulation. Automation of other process steps, including the use of technologies such as robotics, can further reduce risk to the product.
Products should be transferred under appropriate cleanroom conditions. For example, lyophilization processes include transfer of aseptically filled product in partially sealed containers. To prevent contamination, a partially closed sterile product should be transferred only in critical areas.[10] Facility design should ensure that the area between a filling line and the lyophilizer provide for Class 100 (ISO 5) protection. Transport and loading procedures should afford the same protection.
The sterile pharmaceutical drug product and its container-closures should be protected by equipment of suitable design. Carefully designed curtains and rigid plastic shields are among the barriers that can be used in appropriate locations to achieve segregation of the aseptic processing line. Use of an isolator system further enhances product protection .
Due to the interdependence of the various rooms that make up an aseptic processing facility, it is essential to carefully define and control the dynamic interactions permitted between cleanrooms. Use of a double-door or integrated sterilizer helps ensure direct product flow, often from a lower to a higher classified area. Airlocks and interlocking doors will facilitate better control of air balance throughout the aseptic processing facility. Airlocks should be installed between the aseptic manufacturing area entrance and the adjoining unclassified area. Other interfaces such as personnel transitions or material staging areas are appropriate locations for air locks. It is critical to adequately control material (e.g., in-process supplies, equipment, utensils) as it transfers from lesser to higher classified clean areas to prevent the influx of contaminants. For example, written procedures should address how materials are to be introduced into the aseptic processing room to ensure that room conditions remain uncompromised. In this regard, materials should be disinfected according to appropriate procedures or, when used in critical areas, rendered sterile by a suitable method.
If stoppered vials exit an aseptic processing zone or room prior to capping, appropriate assurances should be in place to safeguard the product, such as local protection until completion of the crimping step. Use of devices for on-line detection of improperly seated stoppers can provide additional assurance.
Cleanrooms are normally designed as functional units with specific purposes. The materials of construction of cleanrooms ensure ease of cleaning and sanitizing. Examples of adequate design features include seamless and rounded floor to wall junctions as well as readily accessible corners. Floors, walls, and ceilings should be constructed of smooth, hard surfaces that can be easily cleaned. Ceilings and associated HEPA filter banks should be designed to protect sterile materials from contamination. Cleanrooms also should not contain unnecessary equipment, fixtures, or materials.
Processing equipment and systems should be equipped with sanitary fittings and valves. With rare exceptions, drains are considered inappropriate for classified areas of the aseptic processing facility other than Class 100,000 (ISO 8) areas. It is essential that any drain installed in an aseptic processing facility be of suitable design.
Equipment should be appropriately designed (§ 211.63) to facilitate ease of sterilization. It is also important to ensure ease of installation to facilitate aseptic setup. The effect of equipment design on the cleanroom environment should be addressed. Horizontal surfaces or ledges that accumulate particles should be avoided. Equipment should not obstruct airflow and, in critical areas, its design should not disturb unidirectional airflow.
Deviation or change control systems should address atypical conditions posed by shutdown of air handling systems or other utilities, and the impact of construction activities on facility control. Written procedures should address returning a facility to operating conditions following a shutdown.We have provided over this blog questions and answers for personnel training.
Enzyme linked immunosorbent assay ELISA
http://whoguideline.blogspot.com/2010/04/terminalogy-and-their-explanations.html
Pharmaceutical Aseptic Manufacturing Process Terms , Terminology and Definations.
http://whoguideline.blogspot.com/2010/02/pharmaceutical-aseptic-manufacturing.html
What is CFR 21 PART 11
What is a HEPA filter
Here are some articles which will be useful for you in further understanding of aspects of sterile dosage form manufacturing and regulatory affairs and good manufacturing practice in pharmaceutical industry
Types of validations in pharmaceutical manufacturing
Requirements of documents for validation of sterilisation process
How to investigate OOS out of specification results
Determination of Phenol coeeficient of a disinfectant
Time limitations in sterile pharmaceuticals processing
Aspects of validation of manufacturing process in sterile pharmaceuticals
Controling Pyrogens in injectable dosage forms
Media fill run process simulation aspects Validation of Aseptic Process and Sterilisation
New Drug Application (NDA) how to make a New Drug Application (NDA) to US FDA
How to make Investigational New Drug (IND) Application to US FDA
Drug applications submission to us fda Over the counter Drugs OTC drugs
BIOAVAILABILITY AND BIOEQUIVALENCE REQUIREMENTS
Eletronic record in pharmaceutical manufacturing industry
Good manufacturing practice in pharmaceutical industry
Pharmaceutical industry pharmaceutical companies and FDA latest updates
Here is an intresting article on world wide pharmaceutical industry
Article on Pharmaceutical Industry pharmaceutical industry
This Website is a Guide for Pharmaceutical Manufacturing Pharmacy Students Pharmacy Colleges and Pharmacists pharmaceutical companies health care professionals
To Find Pharmaceutical Jobs and make a Pharmaceutical careers see here pharmaceutical companies
——————————————————————————————————
This Guidance is published by US FDA on September 2004
We also recommend our readers to visit US FDA’S website for undated guidance on sterile drug products

{kind=link}
Leave a Reply